The goal of RexMS is to analyse hydrogen deuterium exchange mass spectrometry (HDX-MS) data at the residue level. The package is designed to be used with data from various platforms that have already been process at Spectrum-level. RexMS takes the processed peptide-level data and infers the residue level deuterium uptake. The underlying model is a Bayesian non-parametric model that recasts HDX-MS data analysis as a (latent) change-point detection problem. The unique benefits of this model are the following:
It can provide statistical confidence at the level of residues (e.g. a probability of change)
Borrow statistical power from overlapping peptides
Infer uptake patterns that are hidden at the peptide-level because of averaging
Provide global and per-residue resolution metrics
It can perform single protein analysis, differential analysis and conformational signature analysis (many states/compounds), linking results to downstream functional outcomes
You can build predictive models with the inferred uptakes using partial least squares discriminant analysis (PLS-DA).
You can costumize the model to your specific needs.
Documentation
The official documentation is available at ococrook.github.io/RexMS. We encourage use to look there and in particular the vignettes. We suggest reading through all of the vignettes first so you understand which part of RexMS you need to use.
Installation instructions
Get the latest stable R release from CRAN. Then install RexMS from Bioconductor using the following code:
Note that you cannot currently install from Bioconductor
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("RexMS")To install the development version of RexMS, it’s easiest to use the remotes package.
if (!requireNamespace("remotes", quietly = TRUE)) {
install.packages("remotes")
}
remotes::install_github("ococrook/RexMS")Example
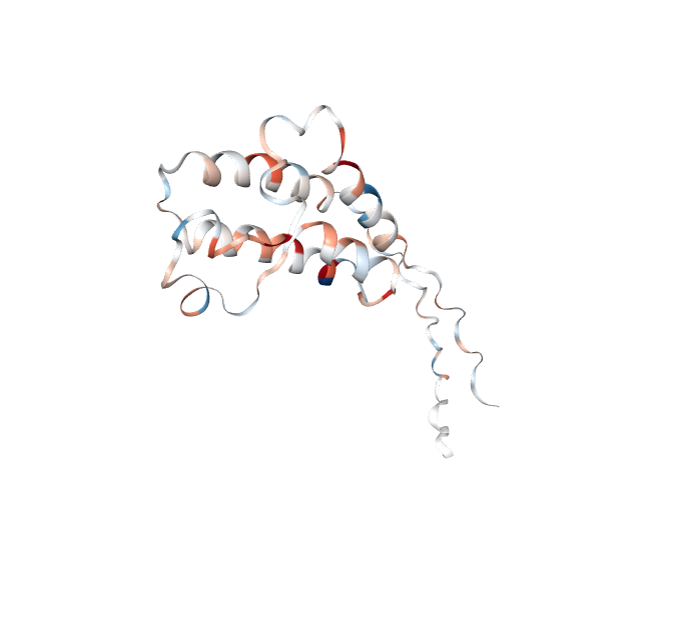
RexMS has structure visualisation at part of its core functionality. Below is an example of how to use the hdx_to_pdb_colours function to map HDX-MS data onto a PDB file.
library("RexMS")
library(NGLVieweR)
# generate random HDX data
v <- matrix(rnorm(n = 477), nrow = 1)
colnames(v) <- seq.int(ncol(v)) # residue numbering
v2 <- v[, seq.int(344, 477), drop = FALSE]
colnames(v2) <- seq.int(ncol(v2))
pdb_filepath <- system.file("extdata", "test_BRD4.pdb", mustWork = TRUE, package = "RexMS")
# generate a protection-deprotection colour mapping
mycolor_parameters <- hdx_to_pdb_colours(v2, pdb = pdb_filepath, cmap_name = "ProtDeprot")
# Note this will open in a view
view_structure(pdb_filepath = pdb_filepath, color_parameters = mycolor_parameters)The expected output is a 3D structure of the protein with the HDX-MS data mapped onto it. The colouring is based on the protection-deprotection scale.The following image is indicative and may not be the same as the output you will see.
Shiny Application
You may be interested in interesting ways to display your data or interact with our results. Here are links to some shiny applications that use RexMS:
https://olivercrook.shinyapps.io/BRD4-ReX/
and
https://olivercrook.shinyapps.io/ConformationalSignatureAnalysis/
Citation
Below is the citation output from using citation('RexMS') in R. Please run this yourself to check for any updates on how to cite RexMS.
print(citation("RexMS"), bibtex = TRUE)
#> To cite package 'RexMS' in publications use:
#>
#> ococrook (2024). _Inferring residue level hydrogen deuterium exchange
#> with ReX_. doi:10.18129/B9.bioc.ReX
#> <https://doi.org/10.18129/B9.bioc.ReX>,
#> https://github.com/ococrook/ReX/ReX - R package version 0.99.2,
#> <http://www.bioconductor.org/packages/ReX>.
#>
#> A BibTeX entry for LaTeX users is
#>
#> @Manual{,
#> title = {Inferring residue level hydrogen deuterium exchange with ReX},
#> author = {{ococrook}},
#> year = {2024},
#> url = {http://www.bioconductor.org/packages/ReX},
#> note = {https://github.com/ococrook/ReX/ReX - R package version 0.99.2},
#> doi = {10.18129/B9.bioc.ReX},
#> }
#>
#> ococrook (2024). "Inferring residue level hydrogen deuterium exchange
#> with ReX." _bioRxiv_. doi:10.1101/TODO
#> <https://doi.org/10.1101/TODO>,
#> <https://www.biorxiv.org/content/10.1101/TODO>.
#>
#> A BibTeX entry for LaTeX users is
#>
#> @Article{,
#> title = {Inferring residue level hydrogen deuterium exchange with ReX},
#> author = {{ococrook}},
#> year = {2024},
#> journal = {bioRxiv},
#> doi = {10.1101/TODO},
#> url = {https://www.biorxiv.org/content/10.1101/TODO},
#> }Please note that the RexMS was only made possible thanks to many other R and bioinformatics software authors, which are cited either in the vignettes and/or the paper(s) describing this package.
Code of Conduct
Please note that the RexMS project is released with a Contributor Code of Conduct. By contributing to this project, you agree to abide by its terms.
Development tools
- Continuous code testing is possible thanks to GitHub actions through usethis, remotes, and rcmdcheck customized to use Bioconductor’s docker containers and BiocCheck.
- Code coverage assessment is possible thanks to codecov and covr.
- The code is styled automatically thanks to styler.
- The documentation is formatted thanks to devtools and roxygen2.
For more details, check the dev directory.
This package was developed using biocthis.






