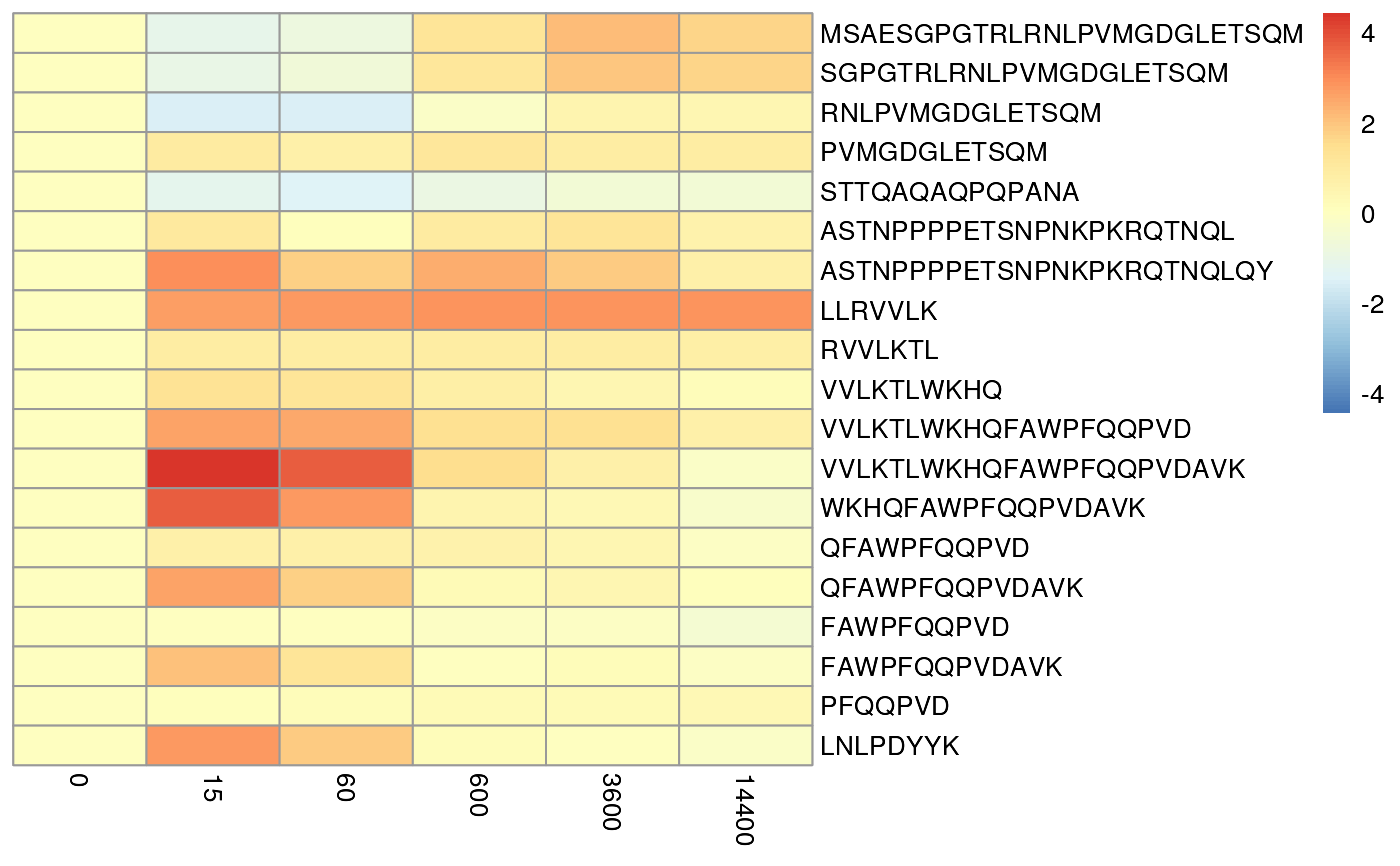
Plot Peptide level error rates
plotPeptideError.RdPlot Peptide level error rates
plotPeptideError(rex_params, HdxData, relative = FALSE)Arguments
- rex_params
An object of class RexParams
- HdxData
An object of class DataFrame, containing the HDX-MS data used.
- relative
Logical, if TRUE the error rates are normalised by the maximum uptake
Value
Returns a pheatmap object
Examples
require(RexMS)
require(dplyr)
data("BRD4_apo")
BRD4_apo <- BRD4_apo %>% filter(End < 100)
numTimepoints <- length(unique(BRD4_apo$Exposure))
Timepoints <- unique(BRD4_apo$Exposure)
numPeptides <- length(unique(BRD4_apo$Sequence))
rex_test <- rex(HdxData = DataFrame(BRD4_apo),
numIter = 10,
R = max(BRD4_apo$End),
numtimepoints = numTimepoints,
timepoints = Timepoints,
seed = 1L,
tCoef = c(0, rep(1, 5)),
BPPARAM = SerialParam())
#> Fold 1 ... Fold 2 ... Fold 3 ... Fold 4 ... Fold 5 ...
#>
|
| | 0%
#> Fold 1 ... Fold 2 ... Fold 3 ... Fold 4 ... Fold 5 ...
#>
|
| | 0%
rex_test <- RexProcess(HdxData = DataFrame(BRD4_apo),
params = rex_test,
range = 5:10,
thin = 1, whichChains = c(1,2))
plotPeptideError(rex_params = rex_test, HdxData = DataFrame(BRD4_apo))